A benefit of working at F2 Labs is that I get to see new and innovative products, straight from the bench of the inventor, and prior to general release. One of our customers, Seymour Duncan, submitted a device for us to work on and I completed the RoHS portion of the project.
I looked at the product offerings on their site during the project and immediately became interested in the after-market pickups they make for guitars. My favorite guitar is the Fender Telecaster because of… well, everything about it. The Telecaster uses a type of pickup that is very articulate but is made from one coil, called a single coil. The result is that it transmits a lot of external ‘noise’ from the surrounding area, called common-mode noise. Things like computers and lighting around the guitar can add unwanted sounds, like loud static and humming. Seymour Duncan makes a replacement set of pickups that are a type of humbucker. Humbuckers are pickups made of two coils (instead of one) which cancel a lot of that humming sound because they are wired together out of phase.
Seymour Duncan Vintage Stack Tele Pickups
Instead of aligning the two rows of coils side by side, these pickups are ‘stacked’ one on top of the other. Hence the name, “Vintage Stack Tele“. The order was placed and within a few days I received these in the mail. Since I work in sales and consulting and not engineering at F2 Labs this seemed like it would be a daunting task. After talking with the Seymour Duncan personnel during our project I learned that it is not hard to do this – and was offered help if I ran into any problems. It turned out to be as easy as he said and all I needed was a $15 soldering iron and some solder.
The Installation!
Next, I took the pick cover from my guitar and, following the included instructions, disassembled the Fender OEM pickups. No turning back now.
Ready for the new pickups.
New, installed Seymour Duncan Vintage Stacks!
Yes, it was as easy as described.
I finished the install and looked at it (and still do) with great satisfaction. Kevin at Seymour Duncan gave me the ‘nudge’ I needed to do this. Thank you Seymour Duncan.
F2 Labs sent a notification to most of our customers regarding some new requirements pertaining to battery cells and battery packs in listed products.
What does this mean?
It means that there are more stringent rules regarding batteries and this will affect your listing if and when you need to revise that listing for any reason. Battery testing can be onerous and expensive. As a side-note, I googled the definition of ‘onerous’ before I used it in this article. See below.
Yes… this fits. We address battery requirements on a daily basis at F2 Labs. That word is an accurate one-word description of the battery testing you will be stuck with if you have planned to use an unapproved or not-suitably-approved battery cell or battery pack in your device. We have had some flexibility to accept batteries in products undergoing US and Canadian Listing Evaluations at F2 Labs in the past. Now OSHA is tightening the requirements, but we can still help you through the process.
Likewise, for CE marking projects, we have some flexibility to accept approved batteries. And we have seen everything from properly approved batteries with test reports to the appropriate standard from accredited labs, batteries with questionable test reports that look like they were drafted in a broom closet, to batteries with no testing whatsoever.
The best time to address this is during the design phase of your project. We call this, “before you start cutting steel,” but you get the idea. Did you know that F2 Labs can provide a design review that can catch potential problems like this prior to sending your device in for testing? We assign an experienced test engineer to review your complete build on paper prior to the build. This can put an end to costly delays to your project by alerting you to unapproved or not suitably approved components, like batteries, in your build before you start making the device.
CONTACT F2 LABS FOR HELP, THIS IS WHAT WE DO.
Want to discuss your project with us?
You can contact us at this link. Our phone number is 877-405-1580 and we are here to help you.
MY SUGGESTIONS AFTER REVIEWING HUNDREDS OFPRODUCTS
F2 Labs assists our customers with compliance issues for many markets across the world. We test equipment in our labs for electrical and mechanical safety & EMC / radio compliance in our three US-based accredited laboratories. But, testing is only part of your compliance process. It means nothing if the rest of your obligations are not met. The minutia of a properly prepared EU declaration of conformity and a correct Technical File with the appropriate documentation are equally important. And RoHS.
If your product has any electric function then chances are that it is in scope of the RoHS Directive 2011/65/EU, including its most important amendment; (EU)2015/863 – commonly called “RoHS 3” or “RoHS 10”. RoHS means restriction of hazardous substances and limits the amount of four heavy metals, two flame retardants, and four phthalates allowed in electrical products.
WHAT RoHS, RoHS 2, RoHS 3, AND RoHS 10 MEAN?
First, let’s define the terminology. RoHS 2002/95/EC was the original RoHS Directive and covered the original six substances (four heavy metals and two flame retardants). RoHS 2002/95/EC was not a CE marking Directive. It was largely ignored and RoHS 2002/95/EC was commonly referred to as “RoHS“.
Next, RoHS 2011/65/EU became effective in 2013 and brought some significant changes. Namely – it was required for CE marking and that includes required reference on the EU declaration of conformity. However, it’s applicability was phased-in and since many types of industrial and medical equipment were part of the phase-in process… it was also largely ignored until about 2017. RoHS 2011/65/EU is commonly referred to as “RoHS 2“.
Finally, many amendments have been added to RoHS 2011/65/EU and most of those deal with specific exemptions to the substance level restrictions. For example, a brass part may contain up to 4% lead content. (EU)2015/863 is the most important amendment and added four additional substances, the phthalates. These are chemicals used in plastics to make the plastic more flexible and soft. They are also dangerous to the human body. The additional phthalate restrictions were enforced starting in July 2019 and, by product category, fully effective on July 22, 2021. RoHS 2011/65/EU combined with (EU)2015/863 is alternately referred to as “RoHS 3” and “RoHS 10“. RoHS 3 because it effectively became the 3rd RoHS and RoHS 10 because it restricts ten substances. However, technically, it is just an amendment to RoHS 2011/65/EU. Therefore a claim of RoHS compliance or even a claim to only RoHS 2011/65/EU is a claim of compliance to RoHS requirements, including all applicable amendments.
Equipment in scope of the RoHS Directive
RoHS COMPLIANCE – A LITTLE MORE DETAIL
We evaluate RoHS compliance for our customers’ products by researching every item on a specially prepared BOM (bill of materials). Every individual item must be included on the BOM and each of these parts are then evaluated for compliance. That includes non-electrical parts like bolts, screws, enclosure materials, plastic parts, labels, and even the paint or coating. This is because if your device is in scope of the RoHS Directive then each part in the build (except a few exceptions like packaging, non-electric consumables, and batteries) must individually comply with the substance level restrictions. Each piece individually.
For example, since lead (Pb) is restricted to 0.1% by weight then each individual piece of your device may not exceed 0.1%. It does not mean that you can use a piece of metal in your device that is 50% lead, but a tiny amount (less than 0.1% of the total weight of your device). For example – if you make a drill press that weighs 1,200 pounds and on the PCB you use a pin that weighs one gram comprised of 0.25% lead… you are not in compliance. 1 gram = 0.002205 pounds. 25% of 0.002205 pounds is 0.00055125. This equals 0.00046%, well below the 0.1% restriction level. But, the restriction level is part-by-part, not against the weight of the whole device. The logic for this is that if you have some substance in a part that is commonly touched, let’s say a button, not a pin buried in the machine on a board, there is a danger to the operator.
THE SUGGESTIONS FOR YOU
Now that we have set up the what and why let’s discuss the how. How do you make sure that by the time your BOM reaches my desk I will go through it and place a check mark next to each part and deliver a final, compliant Technical Report to EN 63000:2018 instead of a discrepancy report showing non-compliant parts?
Suggestion 1
Do not buy any electrical components that are not name-branded. Period. When I have to search for your fuse or cable on a site that also sells cheap socks, bird-food, and coffee filters – and delivers them in the same box – it is almost a certainty that there will be no RoHS compliance documentation, anywhere, for that part. That will stop your project cold.
Suggestion 2
Select for “RoHS compliance” when looking for parts on popular supplier sites like Digikey, Mouser, and Newark. We have no relationship or communication with these three companies.
Suggestion 3
No hardware manufacturer provides RoHS compliance information, that I have seen, like McMaster-Carr. The RoHS information must be collected individually but in my experience it is constantly updated on that company’s website. We have no relationship or communication with McMaster-Carr.
Suggestion 4
Using non-approved assemblies (like motors, power supplies, wireless modules, pumps, etc.) without suitable RoHS compliance will cost you, big. Think of this scenario… you pass all of the electrical and EMC/radio testing performed on your device but the motor in your product has no RoHS compliance information. At this point you are left with a few, equally terrible options:
Pick a new motor, and repeat (at substantial cost) most or all of the compliance testing you just passed.
Pay for a RoHS evaluation on the motor (which will be a completely new evaluation, with the same cost as an evaluation of your entire device… remember, a motor could have hundreds of parts).
Pay for laboratory RoHS testing of the motor. This is not really an option… that could cost $50,000 or $75,000, or more.
Consider this regarding a non-RoHS compliant part that you have tested: you will have to then buy enough parts from that same lot to finish your build because you probably have no control over the materials used in another manufacturer’s device.
Suggestion 5
Specify RoHS compliance, through your purchasing department, for all parts used in any EU build. Catch it on the front-end and you will reduce costly delays on the back-end.
Suggestion 6
We supply very specific instructions for the preparation and submission of your BOM. Please read and follow these instructions. Please call me, directly, with any questions. My direct line is (301)368-2582 and I always answer my phone during business hours unless I am at lunch or I am talking to someone else
CONTACT F2 LABS FOR HELP, THIS IS WHAT WE DO.
Want to discuss your project with us?
You can contact us at this link. Our phone number is 877-405-1580 and we are here to help you.
A lot of our efforts at F2 labs are spent determining the applicability of different regulations to our customers’ products. We help you find out what is applicable and then we suggest the best, fastest, and most cost-effective way to claim compliance. This is a real-life example of how we do it.
Next, see the definition of ‘machinery’ in Article 1., (a). That definition is in Article 2, (a) –
Let’s focus in on Article 2, (a), (5th indent) –
This means that equipment can be in scope of the Machinery Directive if it has moving parts under human power, but only if the function is lifting. So, think of it this way — a bicycle is not in scope, but a hand-cranked hoist for lifting livestock is in scope.
The European Commission sends out a list, every Friday, to show some of the products that have either been stopped in customs or pulled from the market. There are many more products that are pulled, but these are the products categorized as extremely dangerous. I look at this report every week and I look at every mechanical and/or electronic device. I was very interested to read the details of Alert number A12/00729/22 from today.
This is a standard automobile jack that many of us have used. Notice that the alert references EN 1494. That link goes to the Estonian source for EN standards. This organization is highly recommended if you are looking to purchase EN standards. They are always in English and usually for the lowest cost. Yes, you can use EVS-EN 1494 to evaluate a jack for CE conformity to the Machinery Directive for all of the EU – not just for Estonia.
See the scope of EN 1494 below –
Further down in the EN 1494 scope section we see this –
Hmmm. I don’t know about you, but this jack looks exactly like the jack delivered with every vehicle I have owned. This means that if the jack is purpose built for a car manufacturer to deliver with their cars then it does not apply. But, that does not necessarily mean that the Machinery Directive is not applicable. It just means that you cannot use EN 1494 to show conformity with (compliance) to the Machinery Directive.
I would be willing to bet that jacks delivered with new vehicles are under a different regulation. That will not be investigated for this article. Confused? Here is another consideration – if Honda or BMW began buying these jacks for their new cars then those jacks would be excluded from EN 1494, but the very same jacks packaged and re-branded on a shelf in an auto-parts store would be in scope.
This jack manufacturer should have a Technical File that includes an evaluation to EN 1494 since this particular model is in a box and on a shelf, presumably. Also, the only CE marking Directive for this device is the Machinery Directive. RoHS is excluded because there is no electric function. The EU REACH requirement is also applicable, but it is not a CE marking Directive.
IF I HAD TO GUESS – THIS IS WHAT HAPPENED HERE…
Why did this product get flagged? Why are we seeing this alert? If I had to guess (and this is all it is, a guess based on years and years of experience): the jack failed and either property or a person was hurt. The authorities became involved and made a request of the Technical File. Then, (likely) it was discovered that the product was not compliant and there was/is no technical report to show compliance to the Machinery Directive 2006/42/EC, including no report to the applicable harmonized EN standard, EN 1494.
WE CAN HELP YOU TO DETERMINE THE SCOPE OF YOUR PRODUCTS
This is what we do. We can work through the use, application, and construction of your equipment to determine the appropriate compliance path.
Want to discuss your project with us?
We can be contacted via this link. We can be reached by phone at 877-405-1580 and are here to help you.
F2 Labs assists manufacturers around the world with compliance obligations. Historically we have been an accredited test laboratory for electrical, electro-mechanical, EMC, and radio testing. We expanded to offer services to our customers for the EU RoHS Directive 2011/65/EU in 2015 and then added REACH (EC) 1907/2006 as a service offering in 2019.
RoHS and REACH are completely separate compliance requirements and are independent of each other. RoHS is a CE marking requirement and controls the use of ten particular substances. REACH is an EU requirement and, for manufacturers, compels them to identify if any item in their device contains greater than .1% of any of 223 SVHC’s. SVHC means substance of very high concern. We will focus on REACH in this article.
The SVHC list changes every six months. The European Chemicals Agency, also called ECHA, is in charge of this. They never remove substances from this list, it only grows. Last July the list grew from 211 to 219 substances. Four more substances were added in January 2022 to bring it to the current list of 223 SVHCs.
THE WEEDS
Now, let’s not get too far into the weeds. Let’s discuss only the part of REACH that affects companies selling products into the EU. To do that I need to explain a common disconnect between what REACH means, overall, versus what REACH means to you… a business trying to cross the t’s and dot the i’s of EU compliance.
For practical purposes REACH could be two laws… one law that deals with companies that manufacture large amounts of chemicals and another that deals with manufactured items. Lets look at the first scenario; a chemical manufacturer… or an organization that deals with large amounts of chemicals on their own. I thought about an example to use for this to show what I mean and two sprang to mind.
The first example is a company like Dupont. When I think of Dupont the image, below, immediately takes shape.
The above is a train car with thousands of gallons of Titanium Dioxide. I looked up the CAS number (chemical abstract service) for Titanium Dioxide: 13463-67-7. This substance is not on the ECHA SVHC list but it does have compliance requirements under ECHA and REACH: Titanium dioxide ECHA information. Guess what? If you make a hair-dryer or a robotic cell for moving items around this has no bearing on you, at all. It is something Dupont will need to deal with before moving these containers into the EU. By the way, Titanium dioxide is a common chemical, used in things like soap.
The second example is the company in the show Breaking Bad that sold Methylamine to Walt and his colleagues.
That company, I can’t think of its name, bought Methylamine from a German company and then illegally sold it to Walt’s crew. The CAS no. for Methylamine is 74-89-5. It does not appear on the REACH SVHC list. If it is such a dangerous substance then why is it not on the restricted list? Good question.
The answer is that it is controlled by REACH and ECHA — but not in a way that would touch you as a machinery or device manufacturer.
WHAT DOES REACH MEAN TO YOU?
For our customers REACH means something different. For our customers REACH means that you will need to identify if any of the 223 substances of very high concern are in your product. The restriction level is .1% and it applies individually to every part in your machine. For example, lead in the solder on a board in your device will trigger a requirement to alert your customers that your device has SVHC content.
Keep in mind that compliance requirements are triggered once your device exceeds .1% content of any of the (growing) list of 223 SVHCs. The important thing to know is that the weight threshold is not against your device… it is equally applicable to every individual part of your device. So – if we melted that board down and separated it into lead on one side of the balance beam and everything else from the board on the other side there are compliance requirements under REACH if the weight of the lead is greater than .1% of the weight of the whole board.
Now, consider that you will need to do that for all 223 substances of very high concern, individually, for every single item on your BOM, including all the little parts on your board, every screw, the labels, even the paint… and if you continue to sell into the EU you will need to redo this activity every six months!
DON’T THROW YOUR HANDS UP, CALL US!
Throwing your hands up? Don’t. We can handle this for you at F2 Labs and we can do it at the same time we do your RoHS evaluation. Usually we can collect this information from your suppliers. Sometimes quickly, sometimes with a lot of coaching. We take that burden from you. Project by project or every device you sell into the EU. We can even separate your devices by common subassemblies so that if you change a part that is used in multiple SKU’s it can be updated globally in our system – with a simple email to us.
Want to discuss your project with us?
We can be contacted via this link. We can be reached by phone at 877-405-1580 and are here to help you.
We recently fielded a question from a client who had completed all the required testing for their product and asked a question that I’m sure many of you have also asked: “What now?”. The answer, in this case, is to apply all the results of testing we have performed to “certification marks” and decide what we want need to apply to the product or product materials.
International marking regulations can be complex and once you move past the big three (United States, Canada, and Europe) it can, in some cases, be extremely tricky. In this post, I’ll go over some of the more common requirements as well as some specific country cases.
United States and Canadian Safety Listing – in this case, the Nationally Recognized Test Lab (NRTL) that we partner with, is SGS. Their certification mark is used when labeling the product. This mark is heavily controlled, and approval of the associated mark and label is part of the safety approval process. The SGS Q-Mark can only be used once you obtain a successful safety listing report.
FCC (United States) – FCC labeling requirements can differ. They can change, depending on the specifications of the product, on whether it contains unapproved wireless transmitters, and/or on whether it contains a pre-approved wireless transmitter(s). For products that fall under the FCC Supplier’s Declaration of Conformity (SDoC) pathway affixing an FCC label to the product is not a requirement, however, the manufacturer must include a statement in their manual as called out in KDB Publication Number 784748 Appendix A (A.3). For FCC certification, where filing with the commission occurs and an actual FCC ID is provided for the device in question, that same KDB document details the marking requirements under Section 3, and the FCC logo cannot be applied to the finished product.
ISED (Canada) – Pathways to marking a product in Canada mirror the FCC requirements detailed above. For intentional radiators, information for labeling can be found in Section 4 of RSS-Gen Issue 5. Labeling requirements for unintentional products or products that would fall under the SDoC procedure for FCC can be found in Section 5.3.2 of ICES-Gen Issue 1.
CE Certification (Europe) – once you have verified that your product meets the requirements of all applicable directives, you will apply the “CE” mark on the product showing compliance. Here are the requirements as listed on the Europa website, “The CE marking must be visible, legible, and indelible. The CE marking must consist of the initials “CE”, both letters should have the same vertical dimension and be no smaller than 5mm (unless specified differently in the relevant product requirements).”
CB Scheme (International) – There are no labels associated specifically with the CB scheme IEC testing. You will receive a CB test report and CB certificate as deliverables from a successful test. A CB scheme is “a multilateral certification system based on the IEC International Standards.” More information on the CB scheme can be found on the IECEE website.
Japan – products may fall into one of two categories. Category A devices are called “specified devices” and are devices that have a history of accidents in the marketplace. These devices must carry the Diamond PSE mark. Category B devices are called “non-specified electrical products” and are lower-risk products that are subject to a self-declaration scheme like CE. These products must be marked with the Circle PSE mark. For EMC products that fall into the category of multimedia equipment (MME), compliance with Voluntary Control Council for Interference (VCCI) is mandatory. Upon completion, use of the VCCI mark is allowed. Wireless transmitters must obtain MIC certification. More information on the act, testing, and marking requirements can be found on the Ministry of Economy, Trade, and Industry (METI) website.
South Korea – Upon completion of required testing, clients must affix the KC (Korea Certification) to their product. The mark confirms that the product meets the relevant Korean Safety and EMC standards. Korea also has a KS Mark that indicates the product complies with the Korean Industrial Standards (KS). This mark is granted by a certification body and is mostly voluntary. More information about these marks and the requirements for South Korea can be found here.
Australia/New Zealand – Successful completion of required EMC and safety testing will allow the client to place the Regulatory Compliance Mark (RCM) on the product. It signifies compliance with two different schemes: Electrical Equipment Safety System (EESS) and Australian Communications and Media Authority (ACMA). Labeling requirements can be found on the EESS website
South Africa – A certification by the Independent Communications Authority of South Africa (ICASA) provides access to the South African marketplace, specifically for manufacturers of wireless technology. Guidelines for how to affix the ICASA label as well as what it should look like can be found on the ICASA website
Mexico – Norma Oficial Mexicana (NOM) certification applies to any number of up to 2,000 product categories. Once the product is certified to the appropriate NOM safety standard (most of which are based on current IEC standards) and any applicable telecom testing is completed (if the product will connect to the public telecom network) the NOM mark may be applied to the product. Additional information can be found on the Mexican Government’s Website.
F2 Labs can assist your company with worldwide approvals no matter which economy you wish to enter.
This article focuses on the applicability of the Machinery Directive 2006/42/EC and E-Bikes. Since other requirements also apply, they are touched as well below.
We are contacted from time to time by bicycle manufacturers regarding EU and CE requirements for electric bicycles, aka “E-bikes.”. I frequently road-cycle for exercise, and I have looked at E-bikes in my local bike shop for the past year or 18 months. Further, I have seen people riding these and yeah, they look like fun. So, I read, with great interest, that an E-bike was flagged in the EU and the manufacturer was forced to recall them. Imagine the expense of that? Check the alert from the EU’s safety gate, here.
Below is a screenshot of the notification, including the safety issues.
These bicycles are in the scope of the following Directives:
As a quick aside, I want to dedicate just a few words about E-bikes and the RoHS Directive 2011/65/EU + (EU)2015/863. RoHS applies to any product with any electric function, no matter how minor. For example, a lawnmower with a pull-start has just a small electric function but it is in scope. However, there are a few exclusions. See RoHS 2011/65/EU, Article 2, 4. (exclusions), (f):
Article 2, (4.), (f) refers to Regulation (EU) No 168/2013, Article 2., 2. (exclusions), (h):
Since E-bikes are excluded from (EU)168/2013 they cannot be considered as “type-approved” in that regulation. Therefore, they are in the scope of the RoHS Directive and its substance level restrictions. Marking an E-bike with a CE marking is a claim of compliance to the RoHS Directive, intended or not.
Back to the discussion pertaining to E-bikes and the Machinery Directive –
Notice that a specific standard, EN 15194, is referenced. That standard is a C-type Machinery Directive standard. What does that mean? There are three types of Machinery Directive standards –
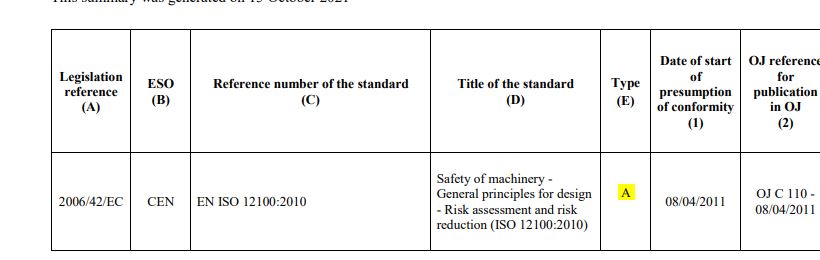
A-type – applicable to all machinery. EN 12100:2010 is the MD’s risk assessment standard and is an example.
B-type – applicable to broad categories of machinery. The main electrical safety standard, EN 60204-1 is an example.
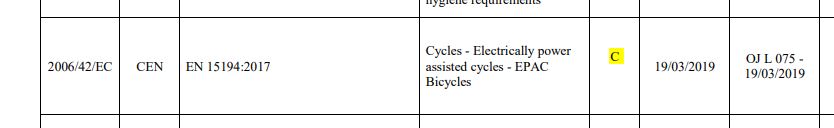
C-type – for a very specific type of machine with unique risks. EN 15194 is an example.
The important thing to understand is that if a C-type standard is applicable to your machinery then that standard (or standards) must be applied to claim compliance to the Machinery Directive. Remember, harmonized standards are voluntary, but they offer the best route to compliance. This is because if you determine the risks from the machinery (Annex I) and then apply harmonized standards to those risks (eliminate/reduce/warn) the resulting EU declaration of conformity will reference the standards used. Equipment that complies with EU Directives by application of harmonized standards benefits from the status of “presumption of conformity.”
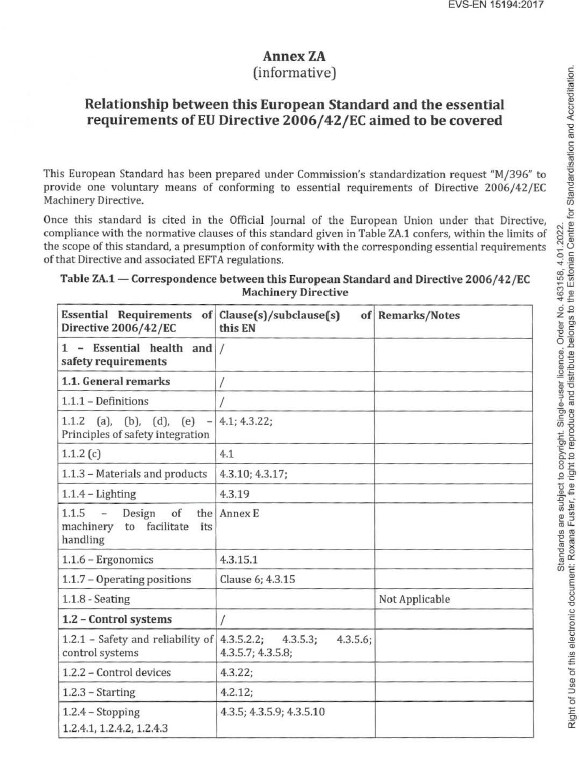
We own a copy of this standard, so I opened it up to look. See the first page (of five) from Annex ZA from EN 15194 below.
Notice that there are several identified Machinery Directive Annex I requirements. I wrote about the relationship between the Annex I requirements and the standards, here: LINK. That article is over two years old but is just as relevant today.
Understanding the table above requires an understanding of the relationship between the harmonized standard (in this case EN 15194) and Annex I of the Machinery Directive. Notice in the table above that (on the left) Annex I EHSR (essential health and safety requirement) 1.1.4 Lighting is referenced. 1.1.4 Lighting is copied from the Machinery Directive and pasted below –
Next, we see that clause 4.3.19 from EN 15194 is listed next to Annex I, 1.1.4 Lighting in the above table from Annex ZA. The table is indicating that if Annex I, 1.1.4 Lighting is applicable (which would be determined during a risk assessment) then the technical requirement for compliance is indicated in clause 4.3.19 of EN 15194.
Clause 4.3.19 sends us to a few ISO standards to address the requirements:
ISO 6742-1 – Cycles – Lighting and retro-reflective devices – Part 1: Lighting and light signaling devices
ISO 6742-2 – Cycles – Lighting and retro-reflective devices – Part 2: Retro-reflective devices
Next, see clause 2 from EN 15194 below. This is the list of additional standards that are attached to the clauses in the table in Annex ZA. In other words, depending upon the outcome of the EN ISO 12100:2010 risk assessment, part of or some of any of these standards may apply.
I looked through EN 15194 extensively this morning. It is filled with requirements that appear to be difficult and complex. For example, braking requirements are littered throughout the standard. It would be very risky to claim compliance to the requirements of this standard without outside technical intervention from an accredited test laboratory. Further, consider what would happen if a person was injured because of poor braking performance? Without a test report or set of test reports from an accredited test lab addressing these and all risks, the consequences could be severe.
Would you know where to start to test the strength of the forks on your E-bike? How about how much distance is required for braking? Or, what about the visibility of the paint or reflectors? All the requirements are in that standard.
Want some help to understand the requirements of CE marking and your EU project? F2 Labs helps manufacturers across the globe every day with small and large products. We have experts who quickly determine what is applicable to the equipment, legally and technically.
We can be contacted via this link. We can be reached by phone at 877-405-1580 and are here to help you.
The CE mark is a mandatory European marking for certain product groups to indicate conformity with the essential requirements set out in European Directives. To use the CE mark on a product, the manufacturer must draw up an EU Declaration of Conformity (DoC) in which the manufacturer attests to conformity with all relevant New Approach Directives (NADs) and takes sole legal responsibility, also called self-certification. In some instances, a NAD may require a Notified Body to issue a Certificate of EU Declaration of Conformity (DoC) to verify the performance of the product or the constancy of the production process (Factory Production Control, for example).
There are numerous directives that products placed on the market in Europe must comply with, however, manufacturers of electronic products have a small list of Directives that are typical, depending on product type.
Radio Equipment Directive 2014/53/EU (RED) – any product that contains a radio module or utilizes wireless technology will more than likely fall into this directive for compliance. “Radio equipment”, as defined in the new RED specifies: ‘radio equipment’ means an electrical or electronic product, which intentionally emits and/or receives radio waves for the purpose of radio communication and/or radiodetermination, or an electrical or electronic product which must be completed with an accessory, such as antenna, so as to intentionally emit and/or receive radio waves for the purpose of radio communication and/or radiodetermination
EMC Directive 2014/30/EU – this directive deals with ensuring that equipment functions satisfactorily in its electromagnetic environment without introducing unacceptable electromagnetic disturbances to other equipment in that environment. The directive ensures the correct operation of equipment that responds to electromagnetic phenomena, is operated in the same electromagnetic environment, and the avoidance of any interference effects.
Low Voltage Directive 2014/35/EU – electrical products that have a voltage rating within the below parameters can be subject to the LVD:
50-1000 volts for AC devices
75-1500 volts for DC devices
Conformity to the LVD is mandatory if your product is within the voltage limits and not excluded by the law. LVD compliance is not assured by using all CE-marked components. That is the starting point and the device itself must carry a CE mark that indicates the overall assembly, tested and evaluated as a single unit, is in legal compliance with the Low Voltage Directive.
Machinery Directive 2006/42/EC – equipment that may fall into the Machinery directive includes machinery, interchangeable equipment, safety components, lifting accessories, chains, ropes, and webbing, removable mechanical transmission devices, and partly completed machinery. The Machinery Directive is a law and requires that equipment within its scope is compliant with the applicable technical requirements listed in Annex I. These technical requirements are called the Essential Health and Safety Requirements (EHSR’s).
RoHS Directive 2011/65/EU – The Restriction of Hazardous Substances (RoHS) Directive is a CE marking Directive that applies to anything that is “EEE” or electrical and electronic equipment, essentially anything that has an electric function, even a gas power with a piezo spark ignitor. Products that fall under the scope of RoHS are found in Annex I of the RoHS Directive and those material restrictions that manufacturers must comply with are found in Annex II. If your product is in the scope of the RoHS Directive, every individual component of said product (paint, screws, housings, labels, etc) must be independently compliant to the RoHS Directive.
Please contact F2 Labs if you have any questions regarding your product’s need for CE compliance.
For years, more niche product categories have suffered from not having harmonized testing standards. What this means for manufacturers is more expensive and extensive regulatory exercises because the standards for the US, Canada, and the European Union are all different.
Now, when new standards are created or adopted, an effort is being made to move towards this harmonization process. Recently, UL 60335-2-40/CSA C22.2 No. 60335-2-40 were adopted and are harmonized with EN 60335-2-40 and IEC 60335-2-40. This UL & CSA certification testing standard applies to multiple products including packaged air conditioners and heat pumps, as well as liquid chillers, dehumidifiers, room air conditioners, and others. This new standard will eventually replace several standards that are used in the HVAC industry, the biggest of which is UL 1995, but will also replace UL 484 and UL 474.
The new standard includes the type testing for each of the tests outlined in UL 1995, however, there are now more extensive requirements and unique tests that are different from what has been done in the past. These include but are not limited to Electronic Fault Test, Glow Wire Test, Flammability Requirements, and additional assessment for power input and current, heating, abnormal temps, ball pressure, amongst others.
The new standard doesn’t go “live” until 2024, and manufacturers may continue to utilize UL 1995 and other appropriate standards for new and existing products but should begin looking at what 60335-2-40 will require of them from a testing standpoint. Manufacturers may begin utilizing 60335-2-40, if they so desire, to get a “head-start” on the competition. However, understand that during this transition period additional changes may be made to the standard as it continues to evolve. Products currently listed to UL 1995, UL 484, UL 474, or similar standards, will more than likely need to undergo full retesting, as UL 60335-2-40 has different test-based requirements as well as new component requirements.
When utilizing the new standard, especially during the transition period, it’s important to have a trusted partner who can help walk you through and effectively guide your team through this process. Reach out to F2 Labs today for Product Safety Testing Services and let us know how we can assist your team in navigating the compliance maze and get your product to market!
Bluetooth and Wi-Fi are technologies that allow our devices to be interconnected, whether that be wireless earbuds, cellular phones, hands-free sets for our cars, tablets, computers, etc. In the United States, products that contain Bluetooth and/or Wi-Fi modules have two pathways towards FCC approval, and manufacturers are legally required to comply with one of these pathways, the details of which are found in CFR 47/Chapter 1/Subchapter A/Part 2/Subpart J.
FCC Certification
The first (2.907 – Subpart J) is what is known as product certification where a finished product, Bluetooth module or Wi-Fi module, etc. may contain an unlicensed or uncertified module, or be, unlicensed or uncertified. In this scenario, we would test the product, Bluetooth module, or Wi-Fi module to FCC Part 15 Subpart C subpart 15.247 or FCC Subpart C subpart 15.249 and produce test results. These test reports would then go through what is called the TCB Review Process (Telecommunication Certification Body) where the reports and product details are reviewed and certification is granted (assuming a passing review), allowing the manufacturer to place an FCC ID on the finished product or Bluetooth module. This ID must be visible on the product whether it is a label, etched on the exterior of the product, or electronically displayed. See more on the FCC rules for labeling here. For more information on Modular approval, requirements click here.
FCC Supplier’s Declaration of Conformity
The second (2.906 – Subpart J) is what is known as a Supplier’s Declaration of Conformity. In this scenario, a product may contain a pre-certified Bluetooth module, or Wi-Fi module, etc. that has been integrated into the host device without making any modifications to the module grant conditions (such as antenna type, gain, power, etc.). Under those conditions, a manufacturer would test for Radiated and/or Conducted Emissions (depending on how the product is powered) under FCC Part 15 Subpart B, 15.107 and/or 15.109 to ensure that the product complies. With a test report in hand and a Supplier’s Declaration of Conformity, the manufacturer is not required to file with the FCC. In this case, the manufacturer would need to place a label on the outside of the product that says it contains the FCC ID of the pre-certified wireless transmitter (i.e., Bluetooth or Wi-Fi module).
F2 Labs is an expert in navigating this sometimes complicated, and confusing process. Reach out to us today, so we can assist you in navigating the compliance maze and getting your product to the market as quickly, correctly, and as efficiently as possible.