This article focuses on the applicability of the Machinery Directive 2006/42/EC and E-Bikes. Since other requirements also apply, they are touched as well below.
We are contacted from time to time by bicycle manufacturers regarding EU and CE requirements for electric bicycles, aka “E-bikes.”. I frequently road-cycle for exercise, and I have looked at E-bikes in my local bike shop for the past year or 18 months. Further, I have seen people riding these and yeah, they look like fun.
So, I read, with great interest, that an E-bike was flagged in the EU and the manufacturer was forced to recall them. Imagine the expense of that? Check the alert from the EU’s safety gate, here.
Below is a screenshot of the notification, including the safety issues.

These bicycles are in the scope of the following Directives:
- Machinery Directive 2006/42/EC
- Electromagnetic Compatibility Directive 2014/30/EU
- Restriction of Hazardous Substances Directive (RoHS) 2011/65/EU + (EU)2015/863
As a quick aside, I want to dedicate just a few words about E-bikes and the RoHS Directive 2011/65/EU + (EU)2015/863. RoHS applies to any product with any electric function, no matter how minor. For example, a lawnmower with a pull-start has just a small electric function but it is in scope. However, there are a few exclusions. See RoHS 2011/65/EU, Article 2, 4. (exclusions), (f):

Article 2, (4.), (f) refers to Regulation (EU) No 168/2013, Article 2., 2. (exclusions), (h):

Since E-bikes are excluded from (EU)168/2013 they cannot be considered as “type-approved” in that regulation. Therefore, they are in the scope of the RoHS Directive and its substance level restrictions. Marking an E-bike with a CE marking is a claim of compliance to the RoHS Directive, intended or not.
Back to the discussion pertaining to E-bikes and the Machinery Directive –
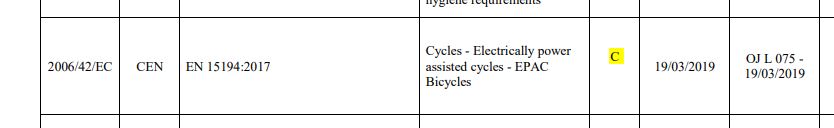
Notice that a specific standard, EN 15194, is referenced. That standard is a C-type Machinery Directive standard. What does that mean? There are three types of Machinery Directive standards –
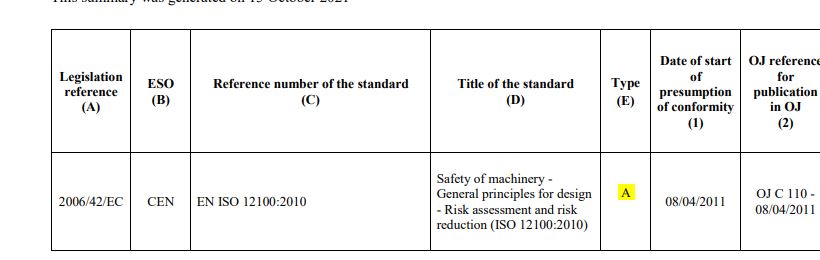
A-type – applicable to all machinery. EN 12100:2010 is the MD’s risk assessment standard and is an example.
B-type – applicable to broad categories of machinery. The main electrical safety standard, EN 60204-1 is an example.

C-type – for a very specific type of machine with unique risks. EN 15194 is an example.
The important thing to understand is that if a C-type standard is applicable to your machinery then that standard (or standards) must be applied to claim compliance to the Machinery Directive. Remember, harmonized standards are voluntary, but they offer the best route to compliance. This is because if you determine the risks from the machinery (Annex I) and then apply harmonized standards to those risks (eliminate/reduce/warn) the resulting EU declaration of conformity will reference the standards used. Equipment that complies with EU Directives by application of harmonized standards benefits from the status of “presumption of conformity.”
See Article 7, and particularly point 2., from the Machinery Directive 2006/42/EC.

The list of standards is published in the Official Journal of the EU and updated periodically at this LINK.
We own a copy of this standard, so I opened it up to look. See the first page (of five) from Annex ZA from EN 15194 below.
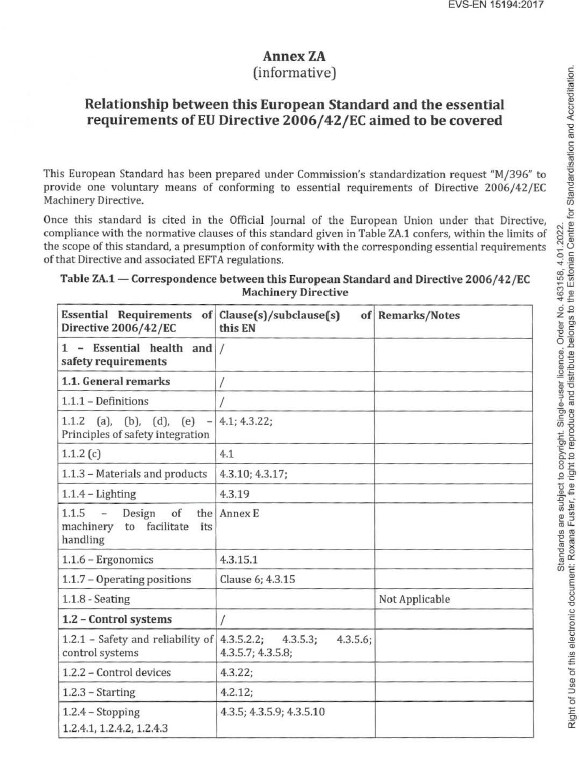
Notice that there are several identified Machinery Directive Annex I requirements. I wrote about the relationship between the Annex I requirements and the standards, here: LINK. That article is over two years old but is just as relevant today.
Understanding the table above requires an understanding of the relationship between the harmonized standard (in this case EN 15194) and Annex I of the Machinery Directive. Notice in the table above that (on the left) Annex I EHSR (essential health and safety requirement) 1.1.4 Lighting is referenced. 1.1.4 Lighting is copied from the Machinery Directive and pasted below –

Next, we see that clause 4.3.19 from EN 15194 is listed next to Annex I, 1.1.4 Lighting in the above table from Annex ZA. The table is indicating that if Annex I, 1.1.4 Lighting is applicable (which would be determined during a risk assessment) then the technical requirement for compliance is indicated in clause 4.3.19 of EN 15194.

Clause 4.3.19 sends us to a few ISO standards to address the requirements:
- ISO 6742-1 – Cycles – Lighting and retro-reflective devices – Part 1: Lighting and light signaling devices
- ISO 6742-2 – Cycles – Lighting and retro-reflective devices – Part 2: Retro-reflective devices
Next, see clause 2 from EN 15194 below. This is the list of additional standards that are attached to the clauses in the table in Annex ZA. In other words, depending upon the outcome of the EN ISO 12100:2010 risk assessment, part of or some of any of these standards may apply.


I looked through EN 15194 extensively this morning. It is filled with requirements that appear to be difficult and complex. For example, braking requirements are littered throughout the standard. It would be very risky to claim compliance to the requirements of this standard without outside technical intervention from an accredited test laboratory. Further, consider what would happen if a person was injured because of poor braking performance? Without a test report or set of test reports from an accredited test lab addressing these and all risks, the consequences could be severe.
Would you know where to start to test the strength of the forks on your E-bike? How about how much distance is required for braking? Or, what about the visibility of the paint or reflectors? All the requirements are in that standard.
Want some help to understand the requirements of CE marking and your EU project? F2 Labs helps manufacturers across the globe every day with small and large products. We have experts who quickly determine what is applicable to the equipment, legally and technically.
We can be contacted via this link. We can be reached by phone at 877-405-1580 and are here to help you.
F2 Labs is here to help.